
~Use this protocol after DNA Extraction~
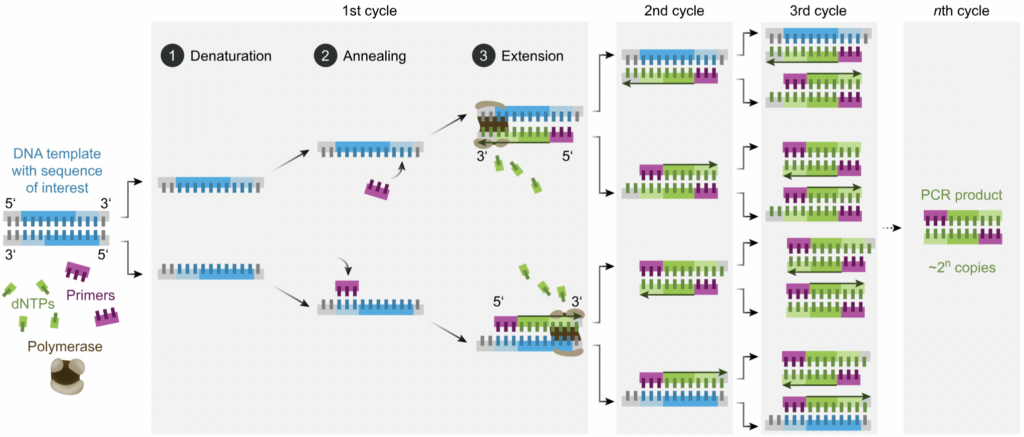
Overview: PCR tests are vital in the use of detecting the presence of genetic material from a specific organism, in our case Arthropods. PCR tests have the ability to amplify a specific segment of DNA as it replicates the segment, making it easier to analyze. This process consists of using a master mix of reagents that help in the isolation and purification of the DNA. The buffer solution in this master mix is vital in maintaining a stable pH through the reaction, and the primers, short DNA sequences, serve as starter DNA sequences for the DNA sequences we are testing to attach onto. After this master mix is created, we add the DNA to the PCR plate to combine the two solutions.
Once our plate is created with a mix of DNA and the master mix, the PCR plate we created undergoes a series of temperature cycles in order to amplify the DNA sequence. The high temperatures in the beginning of the process separates the strands of DNA. This is essential for making the DNA single stranded and accessible. The lower temperatures then allow the primers to bind to their complementary sequences. Lastly, the temperature is slightly raised to extend the primers along the DNA and create new double stranded DNA molecules. Overall, these repeated steps result in the exponential amplification of our targeted DNA sequence which creates many identical copies of the original DNA fragment and allows us to visualize the particular sequence we are looking for.
Technical Protocol
- Each 3 plates that go through TruSeq PCR should receive a different in-line barcode, so that they can be combined later for a sequencing run
- Note in-line barcode letter (A, B, or C) for each plate
| Plate | MCO barcode | ARF1 barcode |
| Plate 1 | MCO_A | ARF1_A |
| Plate 2 | MCO_B | ARF1_B |
| Plate 3 | MCO_C | ARF1_C |
- For each plate of gDNA, 2 TS PCRs will be run- one for the corresponding ARF1 (A, B, or C) and one for the corresponding MCO (A, B, or C) markers.
- Ex: HDI_BE_1 gDNA plate → HDI_BE_1 ARF1a TS PCR
- → HDI_BE_1 MCOa TS PCR
- Ex: HDI_BE_1 gDNA plate → HDI_BE_1 ARF1a TS PCR
- Create plate map: Use the gDNA quantification sheet and sample names to create a plate layout.
- Clean workspace: Put on gloves. Sterilize bench by wiping down with 10% bleach and/or ethanol. Sterilize pipettes with a dilute bleach and then an ethanol wipe-down.
- Calculate master mix: Calculate reagent volumes using the count of samples in plate layout and the table below. Make sure to add one extra for the negative control (nc) and ~2-5 extra to the count for the master mix calculation (even number makes it easier). Remove sufficient reagent volumes to thaw on bench top in tube rack.
| Reagent | Volume (uL) | x100 for 96 well plate | x____ |
| H20 | 2 | 200 | |
| Q reagent | 1 | 100 | |
| F (MCO or ARF1) | 0.5 | 50 | |
| R (fol deg) | 0.5 | 50 | |
| MM | 5 | 500 | |
| Total volume: | 9 |
- Label semi-skirted (preferable) or non-skirted PCR plate:
- Top and side of plate:
- Your name or initials, date, plate name, marker (ARF1 or MCO), inline barcode (A, B, or C), and “TS PCR”
- Surface of plate:
- Indicate empty wells and negative control on plate map and on PCR plate. Use a fine sharpie to outline and mark or tape over empty wells.
- Top and side of plate:
- Vortex and spin: Prepare an ice bucket or use the cold rack from the freezer. Once reagents are thawed, vortex, spin down, and nest in the ice bucket. Likewise, once the gDNA is defrosted, vortex, spin, and put in a plate rack, on ice.
- Make and dispense master mix:
- Combine reagents for the master mix in a labeled 1.5mL tube in the ice bucket. Vortex and spin master mix.
- Pipette 9uL of master mix into each well of the labeled PCR plate that is not taped off/marked as empty. You can choose which kind of pipette to use for this step. It may be easiest to use the repeater pipette with a 0.5mL tip to dispense the master mix.
- Extra reagents can now be frozen until later use.
- If using heat plate sealing, remember to turn on plate sealer. It takes ~ 5 min to heat up.
- Add gDNA: Transfer 1uL gDNA to the PCR plate using a p10 multi-channel pipette. Each sample from the gDNA plate should be transferred to the corresponding well in the PCR plate.
- Seal plate: If using heat seal, walk prepared PCR plate in a plate rack and a non-adhesive aluminum topper to heat sealer. The flashing light indicates the heat sealer is ready. Use heat sealer to seal plate by pressing down for two seconds (one onethousand, two onethousand) – pressing with a bit of a wiggle to get the edges of the plate is very important. Use hand/arm or roller to press on plate seal while still warm to make sure every well is sealed. You should see the small rings of the wells slightly flattened against the aluminum top. Note: It especially fails to seal in row A which leads to evaporation. Remember to turn it off when done. If using other plat sealing methods, take appropriate measure to ensure all wells are sealed.
- Launch thermocycler: Set up thermocycler program to match the table below. Launch program. Collect samples in 2hrs 58mins or later. The last step of the program is an infinite hold at 12 deg C so the plates can be left in the thermocycler for longer if needed.
| Lid 105C, vol 10uL | ||
| Step | Temperature | Time |
| 1 | 95C | 15:00 |
| 2 | 94C | 0:30 |
| 3 | 46C | 1:30 |
| 4 | 72C | 1:30 |
| 5 | Return to step2, 32X | |
| 6 | 72C | 10:00 |
| 7 | 12C | ∞ |
- Next Steps: Preparing for index PCR:
- Run check gel: Use gel electrophoresis protocol to check that you have a bright band at 600bp (ARF1) and 500 bp (MCO). And that there is no amplification in the negative control well.
- Pool samples: Combine MCO_A/B/C plates, by well, including the TS PCR negative controls. You should use the strength of the band of DNA that is visualized on the gel to determine what the relative concentration of DNA is in each well before you pool the wells together. If you have done PCR replicates (e.g., inline_A, inline_B) you will pool these together because they can be distinguished bioinformatically later after demultiplexing based on the indexing combination.
- Next: follow Index PCR Protocol